|
PCR、qPCR、dPCR、RT – PCR、RT-qPCR、Real-Time PCR如何区分?
自从PCR技术被浪出来:PCR传奇史:“浪”出来的生物技术,经历了飞速的发展和技术更新,新冠的洗礼,让所有人都了解到了“核酸检测”的技术。
然而,对于初学者来说,PCR技术的名称和原理往往令人感到困惑,傻傻分不清。某个小伙伴尝试给让你搞懂PCR、qPCR、dPCR、RT - PCR 、RT-qPCR、Real-Time PCR之间的区别和联系。
如果你已经完全分清了,建议继续:多重PCR——病原体检测体系设计原则
1. 普通PCR(就是只做核酸的扩增哦!!!)
聚合酶链反应(Polymerase Chain Reaction,PCR),即一代PCR,是1985年由美国PE-Cetus公司的科学家Kary Banks Mulis发明的一种可在体外快速扩增特定基因或DNA序列的技术。其原理类似于DNA的天然复制过程,在体外条件下以指数级速度扩增,并通过琼脂糖凝胶电泳对产物进行定性分析。PCR由变性(Denaturation)、退火(Annealing)和延伸(Extension)三个基本反应步骤构成:
①变性:通过加热使模板DNA的双链之间的氢键断裂,双链分开而成为单链。这一步骤通常在94℃至95℃的高温下进行,持续30秒至1分钟,确保双链DNA完全解离。
②退火(复性):当温度降低时,引物与模板DNA中互补的区域结合成杂交分子。退火温度通常低于引物本身的变性温度,一般在50℃至65℃之间,持续30秒至1分钟,确保引物与模板DNA特异性结合。;
③延伸(Extension):在DNA聚合酶、dNTPs(四种脱氧核糖核苷三磷酸)和Mg2+等存在下,DNA聚合酶催化引物按照5′→3′方向延伸,合成出与模板DNA链互补的DNA子链。延伸温度通常在70℃至75℃之间,持续时间根据DNA聚合酶的延伸速度和目的扩增片段的长度而定。
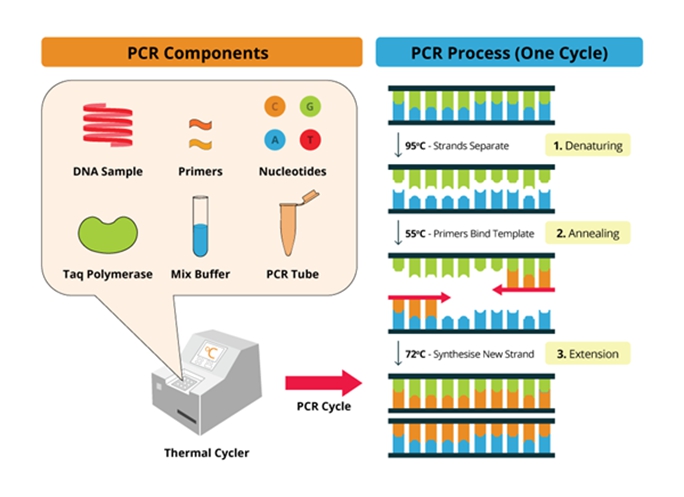
图1 PCR的基本反应原理
如果想用于核酸的检测,其实还需要进行凝胶电泳的辅助。所以很多人认为的第一代PCR,其实更应该像是凝胶电泳技术,而不是PCR技术本身!!!
经过演化和革新,PCR技术已经发展了三代:普通PCR技术(PCR+凝胶电泳)、实时荧光定量PCR技术(qPCR)以及数字PCR(dPCR)技术。
第一代PCR主要缺点:容易发生非特异性扩增和假阳性结果;检测耗时长,操作繁琐;
只能做定性检测。目前,在临床检测中普通PCR已经成为测序等检测技术的前处理方法,没有太多的独立应用空间。在食品药品等检测和分型中,还是不少国家标准的检测技术(毕竟标准的变革不宜那么频繁!!!要加把劲了,跟上新技术。)
2.实时荧光定量PCR(Quantitative Real-time PCR, qPCR)
实时荧光定量PCR,也叫Real-Time PCR,即二代PCR,是指在PCR扩增反应体系中加入荧光染料或者荧光基团,在整个PCR过程中通过收集荧光信号实时监测每一个循环中扩增产物量的变化,最后通过标准曲线和CT值对待测样品进行定量分析。qPCR常用的有两种方法:SYBR Green法和TaqMan探针法。
①荧光染料法(SYBR Green):SYBR Green Ⅰ是荧光定量PCR中最常用的荧光染料,它能与所有的双链DNA结合。在PCR反应体系中,加入SYBR Green Ⅰ,它就会在过程中与双链DNA结合,从而产生荧光信号。因此,反应中发出的全部荧光信号就会与反应中双链DNA的量呈正比,荧光强度也会随着产物的增加而增加。但是由于染料与双链DNA是非特异性结合,如果反应体系中有非特异性扩增或引物二聚体的产生,也将同时被检测,从而可能导致检测结果不准确,因此可能产生假阳性的结果。
优点:价格相对较低;使用方便;检测灵敏度高。
缺点:检测特异性较低,可能产生假阳性结果;无法进行多重【打印本页】【关闭窗口】 |